Projects
An evolutionary-based approach to design the specificity of the Rossmann fold enzymes
PI: Stanisław Dunin-Horkawicz
Foundation for Polish Science grant 5/2018/0014
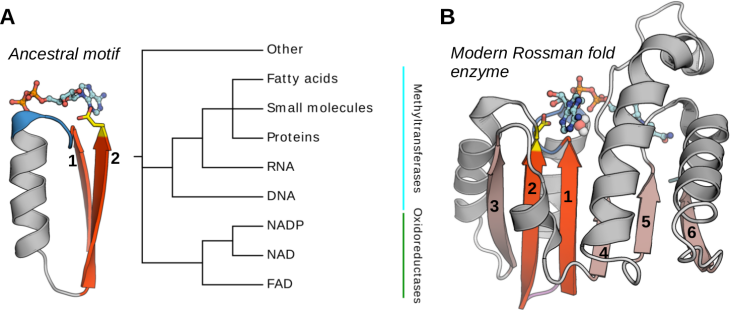
The most general protein similarity is described in terms of protein folds. Protein structures are considered to belong to the same fold if they share the same major secondary structural elements with the same topological connections. It is accepted that the most modern folds evolved via combinatorial shuffling and differentiation events involving a basic set of peptide “building blocks”. Many lines of evidence, including the identification of local sequence and structure similarity within domains of different folds, led to the proposal that the first folded domains arose by repetition, fusion, recombination, and accretion from an ancestral set of functional peptides that emerged in the pre-LUCA RNA world. In modern proteins the descendants of ancestral motifs are indispensable but at the same time they are not independent – their functionality depends on the structural context in which they were embedded in the process of evolution. Considering the above, it becomes evident that research tasks aiming at re-engineering and designing proteins must be performed in the evolutionary context, and that the evolutionary inference must be carried out with the awareness of the complex history of protein folds. Bearing this in mind, we propose to reconstruct the history of the evolution of the Rossmann fold from an ancestral NTP-binding peptide to fully-developed enzymes functioning in modern organisms. Despite its importance, many aspects of the Rossmann fold evolution remain mysterious and a catch-all term “Rossmanoids” is used to avoid evolutionary implications. To fill this gap, we aim to unravel the natural history of the Rossmann enzymes by integrating data from different sources, including sequence, structural, and functional data. This will lead to the development of an evolutionary-oriented classification of all Rossmann enzyme families, which will be an essential tool for investigating Rossmann enzymes and propagating the available experimental data between the families. Rossmann-fold enzymes comprise a clear example of the co-evolution between cofactors and their utilizing enzymes, dating back to the times of pre-LUCA. We, therefore, propose that the understanding of the evolution of the Rossmann fold will reveal the principles that governed the divergence of cofactor specificity of Rossmann enzymes and will open up new possibilities of re-engineering the cofactor specificity and designing new Rossmann-based cofactor binding motifs.
Design of new protein structures with precisely defined features using parametric models
PI: Stanisław Dunin-Horkawicz
National Science Centre grant 2015/18/E/NZ1/00689
Application of the experimental methods revealed that proteins, one of the essential components of the cellular machinery, adopt various structural forms. Proteins consist of combined in various ways, interacting with each other, small building blocks. Most proteins comprise different types of building blocks that are mixed to form compact irregular structures. There are, however, proteins in which one type of building block is repeated continuously, one after the other, resulting in highly regular structures. One of the best-studied structures of this type is the so-called α-helical coiled coils. Regularity of their structures has opened the possibility of developing mathematical models that not only allow describing and quantifying their structural parameters, but also designing completely new structures, not yet observed in nature. Although parametric equations, which constitute a mathematical description of coiled-coil structures, were proposed and implemented already by Francis Crick, they are still the basis for studies aimed at designing new coiled coil-based nanostructures of various applications. The aim of the project described herein is to develop a mathematical model in terms of a set of parameters for two additional classes of repetitive protein structures other than α-helical coiled coils. The first of these types is known as a π-helical coiled coil and is a hypothetical structure, which has not been observed in nature. Its building block, a π-helix, has been studied; however there were no attempts towards amplifying it to obtain a new regular and repetitive structure. The case of the second class of structures, β-helix, is different as many structures of this class have been identified. Development of parametric models for β-helices will enable cataloguing these structures and defining general rules that relate their sequences to structures. This, in turn, will allow designing new β-helix structures, not yet observed in the nature. Accomplishment of this project will be of high importance for studies on protein structures and will lead to creation of a "molecular toolkit" that can be used in designing and creating new structures with precisely defined parameters. Studies on nature-inspired nanomaterials are also important for another reason - they allow validating our understanding of evolutionary processes, which lead to the emergence of the observable "protein universe".
Application of machine learning methods to study bacterial signal transduction systems
PI: Krzysztof Szczepaniak and Aleksander Wiński
Deutscher Akademischer Austauschdienst RISE Worldwide research internship (KS) and National Science Centre “Diamond Grant” DI2018/010748 (AW)
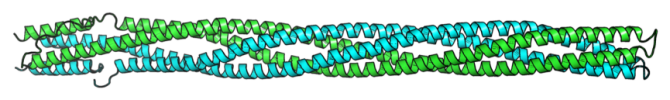
To cope with the need for sensing changes in their inner and outer environments, organisms have evolved signal transduction systems. These systems are the basis for the process of detecting a signal (e.g. nutrients concentration) and coupling this detection with an adaptive cellular response. In prokaryotes, proteins of such signal transduction pathways are frequently built of coiled-coil domains. Coiled-coil domains consist of two or more α-helices wound around each other and forming regular bundles. Signal transduction by these domains relies on the axial rotation of the α-helices and the degree of their rotation can be related to the activity of the downstream effector domains. In our previous studies, we showed that structural parameters of coiled coils can be predicted directly for the sequence information. This prompted us to begin a project that aims at developing a computational tool for prediction of helix axial rotation in coiled-coil domains of bacterial signal transduction proteins. Such a tool will enable us to integrate the available structural, sequence, and functional data, and thus to improve our understanding of bacterial signal transduction systems. In particular, we are interested in comparing coiled-coil signaling domains between various organisms and interpreting the available mutagenesis data.
Molecular mechanisms of the allosteric communication in thymidylate synthase
PI: Jan Ludwiczak
National Science Centre grant 2017/27/N/NZ1/00716
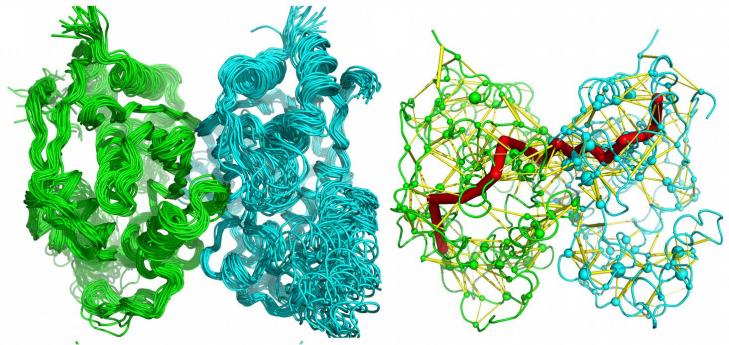
Thymidylate synthase (TS) is a protein enzyme catalyzing an crucial reaction essential for DNA synthesis in all living organisms. Importantly, TS is an established target in chemotherapy with several drugs targeting the enzyme by mimicking either substrate or cofactor (two molecules necessary for assembling DNA building block by the enzyme). Moreover, there is an ongoing research aiming at the creation of selective inhibitors of bacterial and nematode TS variants, which may be used as antibiotics and in treatment of parasitic diseases, respectively. Thymidylate synthase is a homodimer, i. e. it consists of two identical subunits, each containing an active site, a region in protein structure where the reaction occurs. There exists a large body of experimental evidence that TS exhibits “half-the-sites” activity, which means that only one active site is operational at a time. This, in turn, supports the hypothesis of the “molecular communication” between the enzyme active sites. Experimental data seem to support this notion, however only qualitative description for the bacterial enzyme is currently available. Given the fact that such “molecular communication” effects are difficult to investigate with the experimental methods and at the same time are crucial for the understanding of TS functioning, we propose to use computational molecular dynamics simulations. This method is a “microscope” that allows to simulate behavior of a protein structure in solution and to depict, at the molecular level, the dynamical changes. We plan to carry out many simulations of the enzymes from different species with and without bound ligands. This way, we will perform a comparative analysis of the enzyme dynamics and behavior of the active sites. By analyzing the simulated motions of thymidylate synthase structures we hope to identify communication “wires” that connect the two active sites and are responsible for the “half-the-sites” activity. To validate our findings we will perform additional simulations in which the predicted “wires” will be cut. We expect that such “broken” variants of the enzyme will show significantly altered behavior, thus supporting correctness of our predictions. Successful completion of the project will not only allow to significantly increase the knowledge about the dynamics and ”molecular communication” in the thymidylate synthase but also, although this is not the direct goal of the project, will allow to describe the dynamics and subtle differences between the enzymes from different species, which may in future be used to improve existing drugs, or design new that can be used in the targeted therapy against e.g. bacterial infections. Lastly, the outcomes of this project will shed light on the general phenomenon of ”molecular communication” in homodimeric enzymes, which include many, also clinically important, enzymes.
Evolution of protein networks: development of a system to study the evolution of apoptotic machinery
PI: Maja Cieplak-Rotowska and Krzysztof Szczepaniak
Internal CeNT grant DSM/2018
Groups of proteins involved in a specific pathway (e.g. apoptosis) are usually described as “networks”, in which links between proteins signify their direct or indirect interactions and functional coupling. Eukaryotic-like bacterial protein networks (ELB networks) comprise bacterial homologs of eukaryotic proteins involved in innate immunity (Toll-like receptors, NOD-like receptors, and R proteins) and apoptosis (Apaf-1, caspases) [1]. Phylogenetic analysis revealed that eukaryotes acquired functional ELB networks via endosymbiosis and subsequent evolutionary changes resulted in complex networks - as seen in animals such as human, fly or nematode. The function of bacterial ELB systems remains unknown but their features suggest involvement in programmed cell death or/and self versus non-self recognition. This project is a proof-of-concept study towards establishing a simple yeast-based system to study the evolution and function of ELB networks. This research is a collaborative effort that involves Magda Konarska and Maja Cieplak-Rotowska from Laboratory of RNA Biology and Stanisław Dunin-Horkawicz and Krzysztof Szczepaniak from Laboratory of Structural Bioinformatics.